Sickle cell disease is a group of blood disorders affecting hemoglobin, a protein responsible for transporting oxygen in red blood cells. In individuals with the condition, dysfunctional hemoglobin causes red blood cells to become deformed (sickle-shaped). This creates blood flow blockages throughout the body and related serious symptoms, including pain, anemia and infections. The condition also causes damage to organs, including the bones, spleen, liver, brain, lungs, kidneys and joints.1-3
People with sickle cell disease experience sudden and severe episodes of pain as a result of blocked blood flow, which can require medical attention.1,4-6
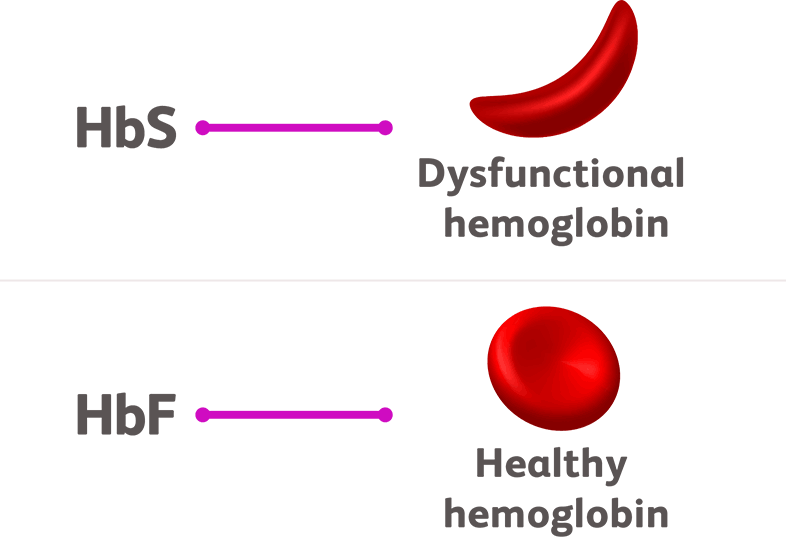
In patients with sickle cell disease, only the HbS form of hemoglobin is dysfunctional. While the HbF form of hemoglobin is unaffected, it is no longer produced in most cases.9
Researchers are investigating ways to revert the body’s production of hemoglobin back to HbF, ultimately increasing the percentage of functional hemoglobin in the body.